作成中。
SNGD法でゲノム編集するためのデザイン(暫定)
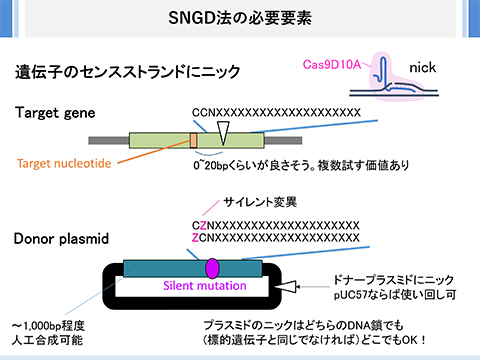
SNGD法による遺伝子編集
SNGD法とは、標的遺伝子に1カ所、ドナープラスミドに1カ所のニックを入れて、ゲノム編集を行う方法です。
Cas9にはspCas9の変異体、Cas9D10Aを用います。
標的遺伝子のセンス鎖にニックを入れる方が良いでしょう。
標的ヌクレオチドの付近にある「CCN(NGG)」配列を見つけ、gRNAのデザインを決定します。Web上にあるフリーデザインツールが便利です。
標的ヌクレオチドがPAM配列あるいは標的配列上になくても大丈夫です。ただ、標的ヌクレオチドとニック部位があまり離れるのは良くありません。だいたい20bpくらいまでがよいかと思います。
ニック部位が少しずれただけでも編集効率が大きく変わる場合があります。可能であれば、いくつか試すのが良いでしょう。
ドナープラスミドには、標的ヌクレオチドの上流・下流を500bp程度含むように、鋳型配列を入れます。ただし、この鋳型配列が、標的遺伝子特異的なCas9-gRNA複合体により切断されてしまってはいけません。
標的ヌクレオチドがPAM配列の(CC/GG)の上にある場合、ドナープラスミド上の鋳型配列は、変えようとする配列そのままで大丈夫です。PAMの直近に標的ヌクレオチドがある場合で、このデザインでいける場合もあります。標的ヌクレオチドがPAM配列から離れている場合、ドナープラスミド上の鋳型配列にはサイレント変異をいれてください。
鋳型配列をpUC57にクローニングする場合には、ドナープラスミドにニックをために既にデザインされたgRNAを利用することが可能です。配列はNakajima
et al. Genome Res. 2018のSupplemental Tableをご参照ください。他のプラスミドの場合、新しくgRNAを設計してください。ドナープラスミドは、人工遺伝子合成を外注するのがよいと思います。おそらく、3~4万円程度で合成してもらえます。シーケンスもばっちりと確認してもらえるので、費用も時間も節約できます。
我々は、いままでCas9D10AとsgRNAを同時発現させることができるプラスミドベクター(Addgene:PX461 or PX462,Dr.
Feng Zhang)を使っています。各プラスミドのブレンド比は、実験ごとに最適化が必要だと思われます。経験的には、PXベクターの2~10倍程度のドナープラスミドがいいと思います。293T細胞で作成した、EGFPcC>Gレポーター細胞にEffectene(キアゲン)でプラスミドをトランスフェクションする場合、24wellフォーマットで、PXプラスミド40~100ng、ドナープラスミド400ngで十分でした。
Cas9を用いたゲノム編集とSNGD法によるゲノム編集
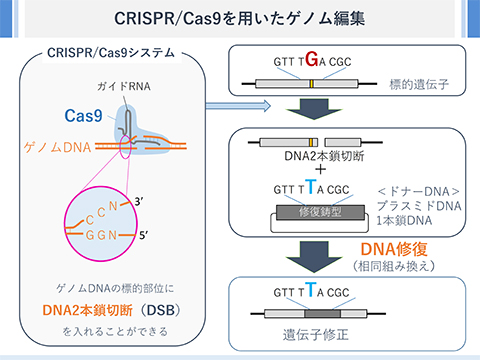
Cas9 はガイドRNAとともに機能し、標的となるDNA配列上にDNA2本鎖切断を発生させます。このDNA切断が、細胞外から導入したドナーDNA(プラスミドDNAや1本鎖DNAが用いられることが多い)配列を鋳型として修復(相同組換え様の機構で修復)されるとゲノムのDNA配列をドナーDNAの改変することができます。図では、「GTT TGA CGC」が「GTT TTA CGC」に書き換えられています。 <<補足>>Cas9はDNA上のPAM配列を認識します。化膿性レンサ球菌由来のCas9(spCas9)の場合、PAM配列はNGGです。DNA配列と相補的なRNA配列を含むガイドRNAによりCas9は標的DNA配列上に固定されます。これにより、ガイドRNAをうまくデザインすれば、狙ったDNA配列上にDNA2本鎖切断を入れることが可能となります。ドナーDNAは、標的とする遺伝子と同じDNA配列を一部含んでいる必要があります。